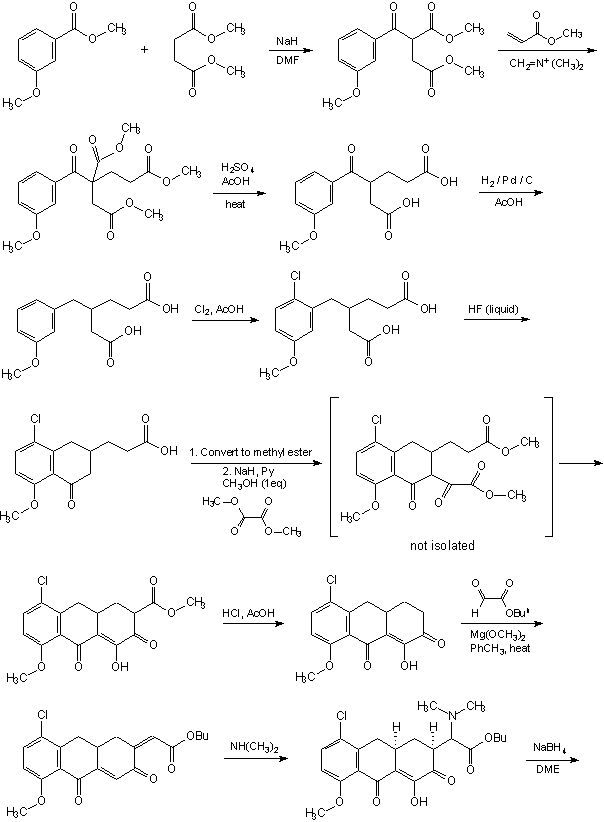
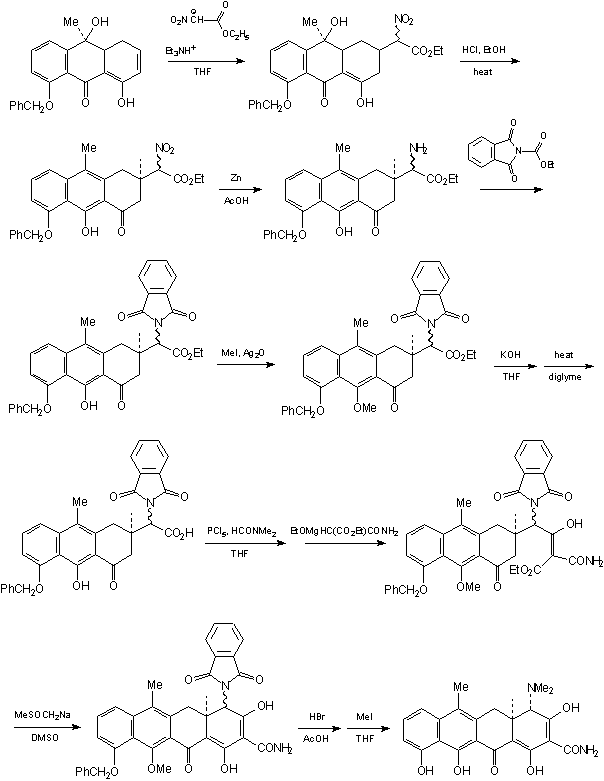
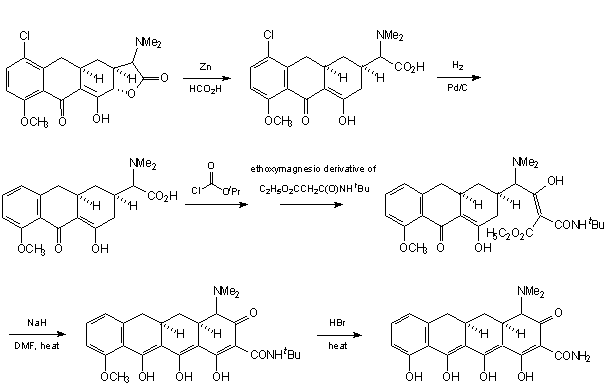
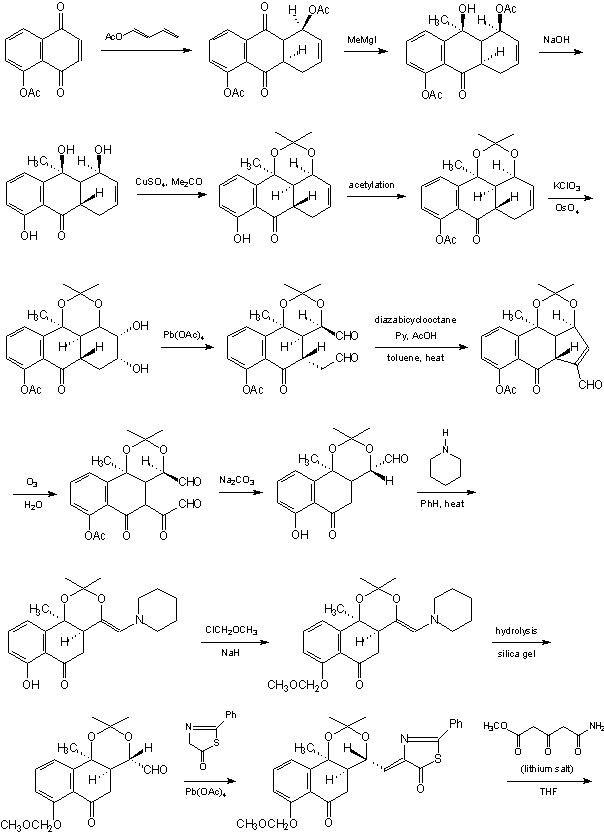
INTRODUCTION
The amounts and identities of the various carotenoids in the photosynthetic membranes of green plants
are relatively well conserved. A handful, typically including lutein, β-carotene, violaxanthin, neoxanthin,
and zeaxanthin, account for most of the carotenoid pigment in the chloroplasts of many plant and
algal species [1]. The biosynthesis and accumulation of these carotenoids in developing chloroplasts
proceed in concert with the assembly of the light-harvesting antennae and reaction centers with which
these pigments are in large part associated [2]. Genetic modifications that reduce or prevent synthesis
of one or more of these carotenoids may be compensated by increases in others so that the total
carotenoid content in the photosynthetic membranes is not much affected [3–7]. Such observations
make clear that robust feedback mechanisms exert control over carotenoid synthesis and accumulation
in plant chloroplasts.
Carotenoid pigmentation in non-green plant plastids, in contrast, ranges broadly both in quantity
and composition. The total amount of the carotenoids may vary from little or none (as in white flower
petals) to quite substantial quantities (as in the dark orange petals of certain marigold varieties). The
pigments may include those common in the photosynthetic membranes (e.g., the lutein of marigold
flower petals), consist of earlier pathway intermediates (e.g., lycopene in red tomato fruits), or be
derived from carotenoids normally found in the chloroplasts (e.g., capsanthin and capsorubin, formed
from violaxanthin in red pepper fruits).
What mechanisms are employed by plants to specify and adjust the amounts and identities of the
various carotenoids that are accumulated in green and non-green plastids? The answer to this question
has many parts, and much remains to be learned. There is abundant evidence to indicate that the reaction
catalyzed by phytoene synthase (PSY) is an important control point for regulation of flux into and
through the carotenoid pathway [8]. This reaction will not be discussed here. Instead, following a brief
update on genes and enzymes of the pathway, I will review what has been learned recently regarding
two other likely control points of the carotenoid pathway in plants: the availability of substrate and
branching of the pathway.
Table 1 Carotenoid pathway genes in Arabidopsis thaliana.
Gene Enzyme Family Members*
Ipi isopentenyl diphosphate isomerase 2
Ggps geranylgeranyl diphosphate synthase 11
Psy phytoene synthase 1
Pds phytoene desaturase 1
Zds ζ-carotene desaturase 1
CrtISO carotene isomerase 1
Ptox plastid terminal oxidase 1
Lcy-b lycopene β-cyclase 1
Lcy-e lycopene ε-cyclase 1
Chy-b β-ring hydroxylase 2
Chy-e ε-ring hydroxylase not identified
Zep zeaxanthin epoxidase 1
Vde violaxanthin de-epoxidase 1
Nsy neoxanthin synthase no ortholog
SUPPLY OF SUBSTRATES (IPP AND DMAPP) FOR CAROTENOID BIOSYNTHESIS
Carotenoids are isoprenoids. The five carbon building blocks that serve as precursors for the synthesis
of carotenoids and other isoprenoid compounds, isopentenyl diphosphate (IPP) and dimethylallyl
diphosphate (DMAPP), are produced in two different compartments and by two different pathways in
plant cells (Fig. 1). The well-known mevalonate (MVA) pathway in the cytosol/endoplasmic reticulum
begins with acetyl-CoA and proceeds in linear fashion to IPP, which is then reversibly converted to
DMAPP in a reaction catalyzed by IPP isomerase (IPI) [15].
The recently recognized methylerythritol (MEP) pathway occurs in plant plastids, in cyanobacteria,
and in certain other bacteria [16; see 17 for a recent update on this incompletely elucidated pathway],
and utilizes pyruvate and glyceraldehyde-3-phosphate (GAP) as the initial substrates (Fig. 1). In
contrast to the MVA pathway, DMAPP and IPP are produced separately via a branching of the MEP
pathway [18]. Even so, IPP isomerase, the enzyme that serves as the terminal enzyme of the cytosolic
MVA pathway (Fig. 1), is also present in plastids [19,20].
Carotenoids in plants are synthesized in the plastids. Are the IPP and DMAPP utilized for
carotenoid synthesis produced solely via the plastid MEP pathway or does the cytosolic MVA pathway
also contribute? Does the source of IPP/DMAPP for plastid isoprenoid synthesis depend on the stage
of development, the type of tissue, or the type of plastid (e.g., etioplast, chloroplast, chromoplast, or
amyloplast)?
Although there are some indications of compartmental “crosstalk”, isoprenoid synthesis in both
green and non-green plastids of many plants has been found to rely primarily on IPP and DMAPP produced
via the MEP pathway. Much of the evidence in support of an MEP pathway origin for plastid isoprenoids
comes from analyses of the distribution of label in certain isoprenoid pathway end-products
after incubation of plants or algae with 13C-labeled glucose or 13C-labeled 1-deoxy-D-xylulose (DOX)
[reviewed in 21]. The effects of specific MVA and MEP pathway inhibitors and the phenotypic consequences
of a mutation in an Arabidopsis gene encoding the MEP pathway enzyme deoxyxylulose-5-
phosphate synthase (DXS) lend further support. The application of the MEP pathway inhibitor fosmidomycin
(an inhibitor of deoxylulose-5-phosphate reductoisomerase, DXR, the first enzyme specific
to the MEP pathway; see Fig. 1) reduces lycopene accumulation in tomato fruits [22], whereas the MVA
© 2002 IUPAC, Pure and Applied Chemistry 74, 1409–1417
Regulation of carotenoid synthesis and accumulation in plants 1411
Fig. 1 Distinct isoprenoid pathways exist in plastids and cytosol of plant cells. Enzymes that may limit flux through
the MEP pathway (the evidence is largely from studies of the bacterium E. coli) are in white text in black boxes.
Abbreviations: diPG, diphosphoglyceraldehyde; GAP, glyceraldehyde-3-phosphate; GAPD, glyceraldehyde-3-
phosphate dehydrogenase; HMG, hydroxymethylglutaryl; MEP, methylerythritol-5-phosphate; PEP, phosphoenolpyruvate.
The amounts and identities of the various carotenoids in the photosynthetic membranes of green plants
are relatively well conserved. A handful, typically including lutein, β-carotene, violaxanthin, neoxanthin,
and zeaxanthin, account for most of the carotenoid pigment in the chloroplasts of many plant and
algal species [1]. The biosynthesis and accumulation of these carotenoids in developing chloroplasts
proceed in concert with the assembly of the light-harvesting antennae and reaction centers with which
these pigments are in large part associated [2]. Genetic modifications that reduce or prevent synthesis
of one or more of these carotenoids may be compensated by increases in others so that the total
carotenoid content in the photosynthetic membranes is not much affected [3–7]. Such observations
make clear that robust feedback mechanisms exert control over carotenoid synthesis and accumulation
in plant chloroplasts.
Carotenoid pigmentation in non-green plant plastids, in contrast, ranges broadly both in quantity
and composition. The total amount of the carotenoids may vary from little or none (as in white flower
petals) to quite substantial quantities (as in the dark orange petals of certain marigold varieties). The
pigments may include those common in the photosynthetic membranes (e.g., the lutein of marigold
flower petals), consist of earlier pathway intermediates (e.g., lycopene in red tomato fruits), or be
derived from carotenoids normally found in the chloroplasts (e.g., capsanthin and capsorubin, formed
from violaxanthin in red pepper fruits).
What mechanisms are employed by plants to specify and adjust the amounts and identities of the
various carotenoids that are accumulated in green and non-green plastids? The answer to this question
has many parts, and much remains to be learned. There is abundant evidence to indicate that the reaction
catalyzed by phytoene synthase (PSY) is an important control point for regulation of flux into and
through the carotenoid pathway [8]. This reaction will not be discussed here. Instead, following a brief
update on genes and enzymes of the pathway, I will review what has been learned recently regarding
two other likely control points of the carotenoid pathway in plants: the availability of substrate and
branching of the pathway.
Table 1 Carotenoid pathway genes in Arabidopsis thaliana.
Gene Enzyme Family Members*
Ipi isopentenyl diphosphate isomerase 2
Ggps geranylgeranyl diphosphate synthase 11
Psy phytoene synthase 1
Pds phytoene desaturase 1
Zds ζ-carotene desaturase 1
CrtISO carotene isomerase 1
Ptox plastid terminal oxidase 1
Lcy-b lycopene β-cyclase 1
Lcy-e lycopene ε-cyclase 1
Chy-b β-ring hydroxylase 2
Chy-e ε-ring hydroxylase not identified
Zep zeaxanthin epoxidase 1
Vde violaxanthin de-epoxidase 1
Nsy neoxanthin synthase no ortholog
SUPPLY OF SUBSTRATES (IPP AND DMAPP) FOR CAROTENOID BIOSYNTHESIS
Carotenoids are isoprenoids. The five carbon building blocks that serve as precursors for the synthesis
of carotenoids and other isoprenoid compounds, isopentenyl diphosphate (IPP) and dimethylallyl
diphosphate (DMAPP), are produced in two different compartments and by two different pathways in
plant cells (Fig. 1). The well-known mevalonate (MVA) pathway in the cytosol/endoplasmic reticulum
begins with acetyl-CoA and proceeds in linear fashion to IPP, which is then reversibly converted to
DMAPP in a reaction catalyzed by IPP isomerase (IPI) [15].
The recently recognized methylerythritol (MEP) pathway occurs in plant plastids, in cyanobacteria,
and in certain other bacteria [16; see 17 for a recent update on this incompletely elucidated pathway],
and utilizes pyruvate and glyceraldehyde-3-phosphate (GAP) as the initial substrates (Fig. 1). In
contrast to the MVA pathway, DMAPP and IPP are produced separately via a branching of the MEP
pathway [18]. Even so, IPP isomerase, the enzyme that serves as the terminal enzyme of the cytosolic
MVA pathway (Fig. 1), is also present in plastids [19,20].
Carotenoids in plants are synthesized in the plastids. Are the IPP and DMAPP utilized for
carotenoid synthesis produced solely via the plastid MEP pathway or does the cytosolic MVA pathway
also contribute? Does the source of IPP/DMAPP for plastid isoprenoid synthesis depend on the stage
of development, the type of tissue, or the type of plastid (e.g., etioplast, chloroplast, chromoplast, or
amyloplast)?
Although there are some indications of compartmental “crosstalk”, isoprenoid synthesis in both
green and non-green plastids of many plants has been found to rely primarily on IPP and DMAPP produced
via the MEP pathway. Much of the evidence in support of an MEP pathway origin for plastid isoprenoids
comes from analyses of the distribution of label in certain isoprenoid pathway end-products
after incubation of plants or algae with 13C-labeled glucose or 13C-labeled 1-deoxy-D-xylulose (DOX)
[reviewed in 21]. The effects of specific MVA and MEP pathway inhibitors and the phenotypic consequences
of a mutation in an Arabidopsis gene encoding the MEP pathway enzyme deoxyxylulose-5-
phosphate synthase (DXS) lend further support. The application of the MEP pathway inhibitor fosmidomycin
(an inhibitor of deoxylulose-5-phosphate reductoisomerase, DXR, the first enzyme specific
to the MEP pathway; see Fig. 1) reduces lycopene accumulation in tomato fruits [22], whereas the MVA
© 2002 IUPAC, Pure and Applied Chemistry 74, 1409–1417
Regulation of carotenoid synthesis and accumulation in plants 1411
Fig. 1 Distinct isoprenoid pathways exist in plastids and cytosol of plant cells. Enzymes that may limit flux through
the MEP pathway (the evidence is largely from studies of the bacterium E. coli) are in white text in black boxes.
Abbreviations: diPG, diphosphoglyceraldehyde; GAP, glyceraldehyde-3-phosphate; GAPD, glyceraldehyde-3-
phosphate dehydrogenase; HMG, hydroxymethylglutaryl; MEP, methylerythritol-5-phosphate; PEP, phosphoenolpyruvate.