| i) The chemical shift of the hydroxyl hydrogen of an alcohol varies with concentration. Very dilute solutions of 2-methyl-2-propanol, (CH3)3COH, in carbon tetrachloride solution display a hydroxyl resonance signal having a relatively high-field chemical shift (< 1.0 δ ). In concentrated solution this signal shifts to a lower field, usually near 2.5 δ. |
| ii) The more acidic hydroxyl group of phenol generates a lower-field resonance signal, which shows a similar concentration dependence to that of alcohols. OH resonance signals for different percent concentrations of phenol in chloroform-d are shown in the following diagram (C-H signals are not shown). |
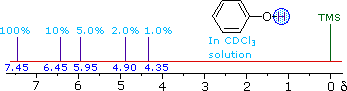 |
|---|
| iii) Because of their favored hydrogen-bonded dimeric association, the hydroxyl proton of carboxylic acids displays a resonance signal significantly down-field of other functions. For a typical acid it appears from 10.0 to 13.0 δ and is often broader than other signals. The spectra shown below for chloroacetic acid (left) and 3,5-dimethylbenzoic acid (right) are examples. |
|
| iv) Intramolecular hydrogen bonds, especially those defining a six-membered ring, generally display a very low-field proton resonance. The case of 4-hydroxypent-3-ene-2-one (the enol tautomer of 2,4-pentanedione) not only illustrates this characteristic, but also provides an instructive example of the sensitivity of the nmr experiment to dynamic change. In the nmr spectrum of the pure liquid, sharp signals from both the keto and enol tautomers are seen, their mole ratio being 4 : 21 (keto tautomer signals are colored purple). Chemical shift assignments for these signals are shown in the shaded box above the spectrum. The chemical shift of the hydrogen-bonded hydroxyl proton is δ 14.5, exceptionally downfield. We conclude, therefore, that the rate at which these tautomers interconvert is slow compared with the inherent time scale of nmr spectroscopy. |
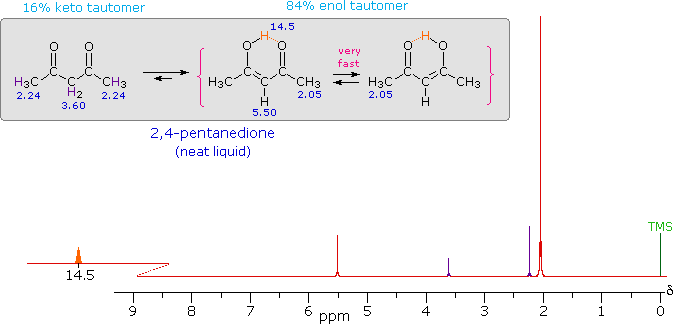 |
|---|
| Two structurally equivalent structures may be drawn for the enol tautomer (in magenta brackets). If these enols were slow to interconvert, we would expect to see two methyl resonance signals associated with each, one from the allylic methyl and one from the methyl ketone. Since only one strong methyl signal is observed, we must conclude that the interconversion of the enols is very fast-so fast that the nmr experiment detects only a single time-averaged methyl group (50% α-keto and 50% allyl). |
No comments:
Post a Comment